


Polyneuropathy is a common neurologic condition with a variety of subtypes that involve the motor, sensory or autonomic fibres.
Distal symmetric polyneuropathy is the most common subtype of polyneuropathy; it classically presents with sensory-predominant, length-dependent symptoms and signs.
Clinical features such as acute-to-subacute onset, asymmetry, non–length dependence, motor-predominant signs and associated systemic features should prompt urgent neuromuscular referral for investigation of polyneuropathy.
Most causes of distal symmetric polyneuropathy can be identified through evaluation of a patient’s medical history; some causes are identified with screening laboratory tests.
First-line oral treatment options for the symptoms of painful diabetic neuropathy are tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors, sodium-channel blockers, and gabapentinoids, which all appear to be similarly effective.
Polyneuropathy is a common neurologic condition with an overall prevalence in the general population of about 1%–3%, increasing to roughly 7% among people older than 65 years.1 Polyneuropathy has many causes, and can present in many different ways; thus, it requires a logical clinical approach for evaluation, diagnosis and management. We review the approach to evaluating a patient with polyneuropathy by highlighting important aspects of the history and neurologic examination. We focus on the role of diagnostic investigations for distal symmetric polyneuropathy (DSP), the most common subtype, and an approach to the symptomatic treatment of painful diabetic polyneuropathy (PDN). We draw on practice-based guidelines, meta-analyses and systematic reviews, where possible, as they represent the highest levels of evidence (Box 1).
Box 1: Search strategy for this review
Using PubMed, we screened all publications from the last 10 years that pertained to the management of polyneuropathy in the primary or urgent care setting, using the search terms “polyneuropathy,” “peripheral neuropathy,” and “management.” We reviewed those that were relevant to the subject, and emphasized practice guidelines, meta-analyses and reviews. For practice guidelines, we extended this search to the last 20 years and reviewed the references of each included guideline for potential inclusion.
What are the clinical features of a polyneuropathy?
Symptoms of polyneuropathy can be categorized by whether they involve sensory, motor or autonomic fibres. Sensory fibres include large-diameter fibres, which mediate vibratory sensation and proprioception, and small-diameter fibres, which mediate pain and temperature sensation. Both modalities should be examined because their relative impairment is a clue to the cause. Dysfunction of either type of sensory fibre can result in sensory alteration, which can range from paresthesias, described as “pins and needles” (positive sensory symptoms), to substantial or complete loss of sensation, known respectively as hypoesthesia and anesthesia (negative sensory symptoms). Large fibre sensory dysfunction may result in gait impairment caused by loss of proprioception (sensory ataxia). Small fibre sensory dysfunction most often causes pain, with some patients having hyperesthesia, an accentuated sensation of tactile stimulation, or allodynia, the perception of normally nonpainful stimuli as painful. Spontaneous episodes of pain can be accompanied by redness and swelling of the affected skin owing to the transmission of unprovoked pain signals by damaged sensory C-fibres, which also release vasoactive substances that cause neurogenic inflammation.2 Additional small-fibre sensory symptoms include deep aching, postexertional malaise and neuropathic itch.3 In contrast, patients with involvement of the motor fibres will primarily describe weakness, which may manifest as loss of dexterity, gait disturbance or both. Autonomic symptoms caused by neuropathy may be underrecognized because these symptoms may have many other causes. Common autonomic symptoms include orthostatic intolerance, gastroparesis, constipation, diarrhea, neurogenic bladder, sexual dysfunction, pupillomotor (i.e., blurry vision) symptoms and vasomotor symptoms, which can lead to dry eyes, mouth or skin, or burning and flushing of the skin.4
Distal symmetric polyneuropathy is the most common subtype of polyneuropathy and is characterized by a length-dependent process whereby the longest nerves are affected first. Findings include symmetric distal weakness with sensory loss (small fibres, large fibres or both) and diminished or absent ankle reflexes.5 Sensory loss begins in the feet, in a nondermatomal, multiple nerve distribution.5 When sensory symptoms or signs reach the upper calf, the fingertips become affected as the nerve lengths in these areas are roughly equivalent; this is known as the glove and stocking pattern of sensory loss.6 In severe cases, this pattern is followed by sensory loss in the midline anterior chest and abdomen, owing to distal degeneration of the thoracic intercostal nerves. Weakness occurs after sensory loss, first affecting toe extension, then ankle dorsiflexion.6 Autonomic symptoms may occur if small sensory fibres are also affected, and typically begin distally, with sweating abnormalities or circulatory instability of the feet.5
Clinicians should be aware of conditions that may mimic polyneuropathy, particularly cervical myelopathy, which can present with a pyramidal distribution of weakness (i.e., preferential weakness of upper limb extensors and lower limb flexors), hyperreflexia below the level of the lesion, a sensory level, and bladder and bowel dysfunction. Acute causes of myelopathy, including cord compression or ischemic infarction, can present with loss of reflexes at and below the level of the lesion.
How should patients be assessed?
An approach to the clinical assessment of a patient with possible polyneuropathy, and a guide for which patients to refer urgently to a specialist, is provided in Figure 1. Patients with certain symptoms and signs require urgent onward referral, and recognizing subtypes of polyneuropathy and important differential diagnoses is important to direct management.

Approach to assessing a patient with suspected polyneuropathy. *Screening positive on ODS increases likelihood of an inflammatory cause or other treatable neuropathy. Note: ODS = onset, distribution and systemic features tool.
Patients with neuropathies with an acute (evolving over days) to subacute (evolving over weeks) onset, with plateauing of symptoms (stability of accrued neurological deficits) within 8 weeks from symptom onset, require urgent referral to a neuromuscular specialist.7 This also applies to peripheral neuropathies with hyperacute onset of symptoms (e.g., wrist or foot drop) as they raise concern for a vasculitic process. Patients with acute neuropathy who have substantial pain or associated trauma may require urgent management via the emergency department.
Additional patterns that are highly suggestive of vasculitic neuropathy include rapidly progressive, painful polyneuropathy and multiple concurrent mononeuropathies (mononeuritis multiplex), where mononeuropathy is defined as signs or symptoms attributable to 1 nerve.8 A non–length-dependent pattern with both proximal and distal weakness may indicate an inflammatory demyelinating process, such as chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), particularly if motor deficits are comparable to or greater than sensory deficits. A less common diabetic neuropathy, diabetic lumbosacral radiculoplexus neuropathy, is a painful, rapidly evolving, asymmetric lower limb neuropathy that can cause severe morbidity.
The differential diagnoses for subtypes of polyneuropathy are listed in Table 1. A patient with diffuse areflexia without other neurologic deficits on neurologic examination, may require referral given the possibility of a hereditary cause. Isolated, asymmetric reflexes, such as the loss of only 1 ankle reflex, may suggest a mononeuropathy or radiculopathy, for which non-urgent referral for neuromuscular evaluation is appropriate. Distal calf atrophy, hammer toes and pes cavus (high-arched feet) are characteristic of a long-standing neuropathy, often seen in hereditary neuropathies (Figure 2).
Differential diagnoses for subtypes of polyneuropathy9
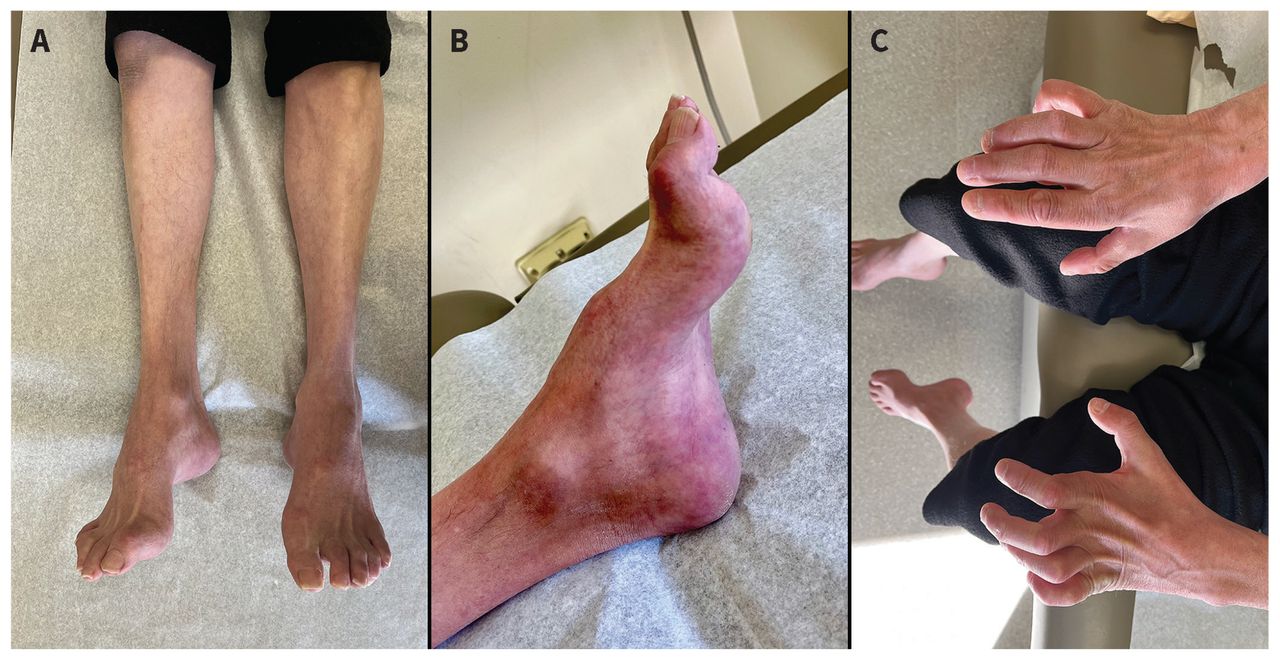
Photographs of a patient with chronic polyneuropathy from Charcot–Marie–Tooth disease, with findings of (A) atrophy of the distal leg and foot musculature; (B) pes cavus (high arch), hammer toes and skin discolouration on the feet; and (C) severe atrophy of intrinsic hand muscles (including the lumbricals) with relative preservation of forearm muscles, resulting in metacarpophalangeal hyperextension and proximal interphalangeal and distal interphalangeal flexion, referred to as “claw hands.”
A diagnostic screening tool using onset, distribution and systemic features can be used to accurately identify patients with inflammatory neuropathies. Patients who screen positive using this tool have 1 or more of the following features: acute or subacute onset (< 8 wk to reach plateau), non–length-dependent distribution or at least 1 systemic sign (skin changes, weight loss, autonomic symptoms, fever and chills, or joint inflammation).7 The screening tool is 96% sensitive and 85% specific in identifying inflammatory neuropathies, including Guillain–Barré syndrome, CIDP (and its subtypes), multifocal motor neuropathies, vasculitic neuropathies and paraneoplastic neuropathies.7 Systemic findings in a patient with an axonal neuropathy would also prompt referral to a neuromuscular specialist. Prompt recognition and treatment is important as prolonged duration of disease without adequate treatment of these neuropathies can lead to serious morbidity.
Electrophysiological studies can assist with diagnosis and guide management and clinical follow-up. In many parts of Canada, it is difficult to access a neuromuscular subspecialist in a timely manner and the patient should be referred to a general neurologist. Patients with suspected Guillain–Barré syndrome should be referred to their local emergency department for evaluation and consideration of admission to the hospital because of the possibility of progressive weakness, respiratory failure and dysautonomia. Urgent electrodiagnostic evaluation is indicated, as testing can confirm the diagnosis and lead to specific and definitive management.10
What tests aid in the diagnosis of distal symmetric polyneuropathy?
The presence of neuropathic symptoms and signs, and of electrodiagnostic findings provide the highest level of accuracy for the diagnosis of DSP.5 Findings on physical examination include decreased or absent ankle reflexes, decreased distal sensation, and distal muscle weakness or atrophy. Neuropathic symptoms alone have relatively poor diagnostic accuracy; signs are better predictors and should be weighed more heavily.5 Electrodiagnostic studies provide a higher level of specificity to the clinical diagnosis.11,12
Whether all patients with suspected DSP should receive electrodiagnostic evaluation is unclear. Four observational studies found that electrodiagnostic evaluation was associated with a change in diagnosis, management or both for more than 40% of patients, but only 1 study was focused on patients referred for DSP.13–16 However, a retrospective cohort study found a change in management in fewer than 1% of patients with DSP seen by community neurologists.17 These disparate findings likely reflect the exclusion of many other types of neuropathies in this study, as more than 80% of study patients had a neuropathy attributed to abnormal glucose metabolism.18
We suggest performing electrodiagnostic evaluation for most patients who present with polyneuropathy, including DSP, especially if the cause is not established. Nerve conduction studies and electromyography allow the electromyographer to categorize the polyneuropathy as primary axonal or demyelinating; motor, sensory or both; or acquired or inherited. All of these categories have important implications for determinining cause and management.9 For example, distal acquired demyelinating symmetric polyneuropathy, a variant of CIDP, presents like DSP but requires relevant investigations for mimics and often requires treatment with immunotherapy.19 Additional valuable information obtained from nerve conduction studies and electromyography includes chronicity, prognosis and, in some circumstances, response to therapy.
Patients with exclusively small-fibre sensory neuropathy will have normal findings on electrodiagnostic studies because these tests evaluate function of only motor and large sensory fibres. Skin biopsy with measurement of the intraepidermal nerve fibre density is a validated, reproducible marker of small-fibre sensory pathology.20 This is the gold standard test for the diagnosis of small-fibre neuropathy, as recommended by the American Academy of Neurology (AAN), with a sensitivity of 45%–90% and specificity of 95%–97%.21 However, its availability and reimbursement vary widely across Canada, thereby limiting its use. The AAN practice guideline also recommends that clinicians consider autonomic testing (i.e., sudomotor, cardiovagal and vasomotor adrenergic testing) in the evaluation of patients with suspected autonomic neuropathies and in those with small-fibre sensory neuropathies.21 Nerve biopsy is useful when vasculitis, sarcoidosis and infiltrative disorders (such as malignant disease or amyloidosis) are suspected.
How is the cause of an acquired distal symmetric polyneuropathy determined?
The history and neurologic examination are paramount in determining the cause of DSP. Overall, nearly 60% of patients will have an identified cause after history and physical examination for DSP; screening laboratory tests can be used to discover a cause in an additional 10% of patients, leaving around 30% of cases as idiopathic.6,22,23 Chronic idiopathic axonal neuropathy is the term reserved for ambulatory patients who have a slowly progressive polyneuropathy, typically after the age of 60 years.24
Common causes of DSP include diabetes, history of alcohol use disorder, renal impairment and medications, including chemotherapy, with diabetes being the most common (Table 2).26 About 40%–50% of patients with diabetes mellitus will develop a detectable neuropathy within 10 years after onset.27 However, other treatable causes for DSP should still be considered among patients with diabetes.27–29 Alcohol-related neuropathy is an irreversible, slowly progressive DSP that is likely mediated by a combination of direct toxic effects and secondary vitamin deficiency, such as a B1 or B12 deficiency.25 Uremic neuropathy occurs in as many as 90% of patients with a glomerular filtration rate below 6 mL/min/1.73 m2.30 Medications and chemotherapy agents can also cause DSP (Table 2).
Common medications associated with polyneuropathy25
Screening laboratory tests are routinely used for patients with DSP to test for treatable causes. The AAN practice guideline recommends high-yield screening laboratory tests for all patients with DSP (summarized in Box 2).31
Box 2: High-yield screening laboratory tests for distal symmetric polyneuropathy
What is the approach to symptomatic treatment of painful neuropathy?
In general, the treatment of chronic neuropathic pain is not influenced by the cause, as limited evidence exists to suggest that specific medications are more effective for specific disorder.32 Notable exceptions include trigeminal neuralgia and cancer-related neuropathic pain. The Special Interest Group on Neuropathic Pain published an international guideline on pharmacotherapy for neuropathic pain.32 A more recent comprehensive guideline from France provides recommendations on pharmacological and nonpharmacological treatment for focal, central or diffuse peripheral neuropathic pain.33
Much of the literature for the symptomatic treatment of painful neuropathy is derived from studies on patients with PDN. Neuropathic pain is one of the most disabling symptoms for these patients.34 It occurs in about 40%–60% of patients with diabetes and neuropathy, yet few patients are treated for pain despite the availability of effective treatments.35,36
The AAN practice guideline on symptomatic treatment of PDN was derived from a meta-analysis that included only randomized controlled trials with a maximum duration of active treatment of 16 weeks.37 Most clinical trials quantified successful treatment as a 30% reduction in pain,37 and few patients achieve more than a 50% reduction with any single drug.38 Patients should therefore be counselled that the goal of therapy is to reduce, not eliminate, pain to align patients’ expectations with the expected efficacy of interventions.38 Evidence from this meta-analysis showed that 4 classes of oral medications reduce pain, namely tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors (SNRIs), sodium-channel blockers and gabapentinoids. The effect sizes were similar among the 4 classes of medications and all are options for first-line monotherapy among patients with PDN.37 Given the similar efficacy of these medications, clinicians should consider their adverse effect profile and cost, as well as patient comorbidities to determine the first-line oral agent.
Opioids can provide short-term pain reduction in patients with PDN, but the evidence that they are effective in the long term is weak.39,40 Opioids and opioids combined with SNRIs are discouraged for long-term management owing to lack of efficacy, high dependence rates and dose-dependent risk of serious adverse effects.27,37,38 Cannabis-based medicines, including nabilone, a synthetic cannabinoid, may help improve neuropathic pain; however, more studies are needed as their benefits may be outweighed by their potential harms.37,41,42
Clinicians should assess the efficacy of first-line therapy after the medication has been titrated to an efficacious dose for 6–12 weeks.37,43 The OPTION-DM trial, a multicentre, randomized, double-blind trial, compared the efficacy of combination therapies (amitriptyline with pregabalin, pregabalin with amitriptyline or duloxetine with pregabalin) for the primary outcome of pain relief and for secondary outcomes (including quality of life, mood and sleep) among patients with PDN. Combination therapies for patients who did not respond to monotherapy at 6 weeks had similar efficacy across primary and secondary outcomes.43 This trial provides evidence for using combination therapy of first-line medications for patients with PDN if they have suboptimal response to monotherapy. It is also important to assess and treat comorbid mood and sleep disorders, and consider topical or nonpharmacological interventions.37
Conclusion
The diagnosis and management of patients who present with polyneuropathy requires a systematic approach. Distal symmetric polyneuropathy, the most common subtype, is characterized by a combination of neuropathic symptoms and findings on physical examination and electrodiagnostic testing. Most causes of DSP can be identified after an evaluation of a patient’s medical history and completion of high-yield screening laboratory tests, but some patients will be classified as idiopathic. Patients with diabetic neuropathic pain should be offered symptomatic pharmacological options, which are effective at reducing pain but not eliminating it. Future research should address key gaps in our current knowledge of symptomatic treatment of neuropathic pain caused by polyneuropathy (Box 3).
Box 3: Unanswered questions
Should all patients with symptoms and signs consistent with distal symmetric polyneuropathy and a known cause be referred for nerve conduction studies and electromyography for diagnostic confirmation?
What is the threshold for additional laboratory testing beyond high-yield screening tests among patients with progressive symptoms and signs of polyneuropathy?
Do all patients with only symptoms of isolated small-fibre neuropathy require a skin biopsy, given its wide differential diagnosis?
What management strategies are best for neuropathic pain caused by polyneuropathy?
Acknowledgement
The authors thank the patient for allowing use of these educational images in this publication.
Footnotes
Competing interests: None declared.
This article has been peer reviewed.
Contributors: Ario Mirian, Ziyad Aljohani, Daniel Grushka and Anita Florendo-Cumbermack contributed to the conception and design, acquisition of data and interpretation of the literature. Ario Mirian drafted the manuscript. All of the authors revised it critically for important intellectual content, gave final approval of the version to be published and agreed to be accountable for all aspects of the work.
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY-NC-ND 4.0) licence, which permits use, distribution and reproduction in any medium, provided that the original publication is properly cited, the use is noncommercial (i.e., research or educational use), and no modifications or adaptations are made. See: https://creativecommons.org/licenses/by-nc-nd/4.0/
References
In this issue

Article tools






{kind=link}
{kind=link}
Jump to section
- Article
- What are the clinical features of a polyneuropathy?
- How should patients be assessed?
- What tests aid in the diagnosis of distal symmetric polyneuropathy?
- How is the cause of an acquired distal symmetric polyneuropathy determined?
- What is the approach to symptomatic treatment of painful neuropathy?
- Conclusion
- Acknowledgement
- Footnotes
- References
- Figures & Tables
- Related Content
- Responses
- Metrics
Related Articles
Cited By...
More in this TOC Section
Similar Articles


Podcast